{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
EMD-20926
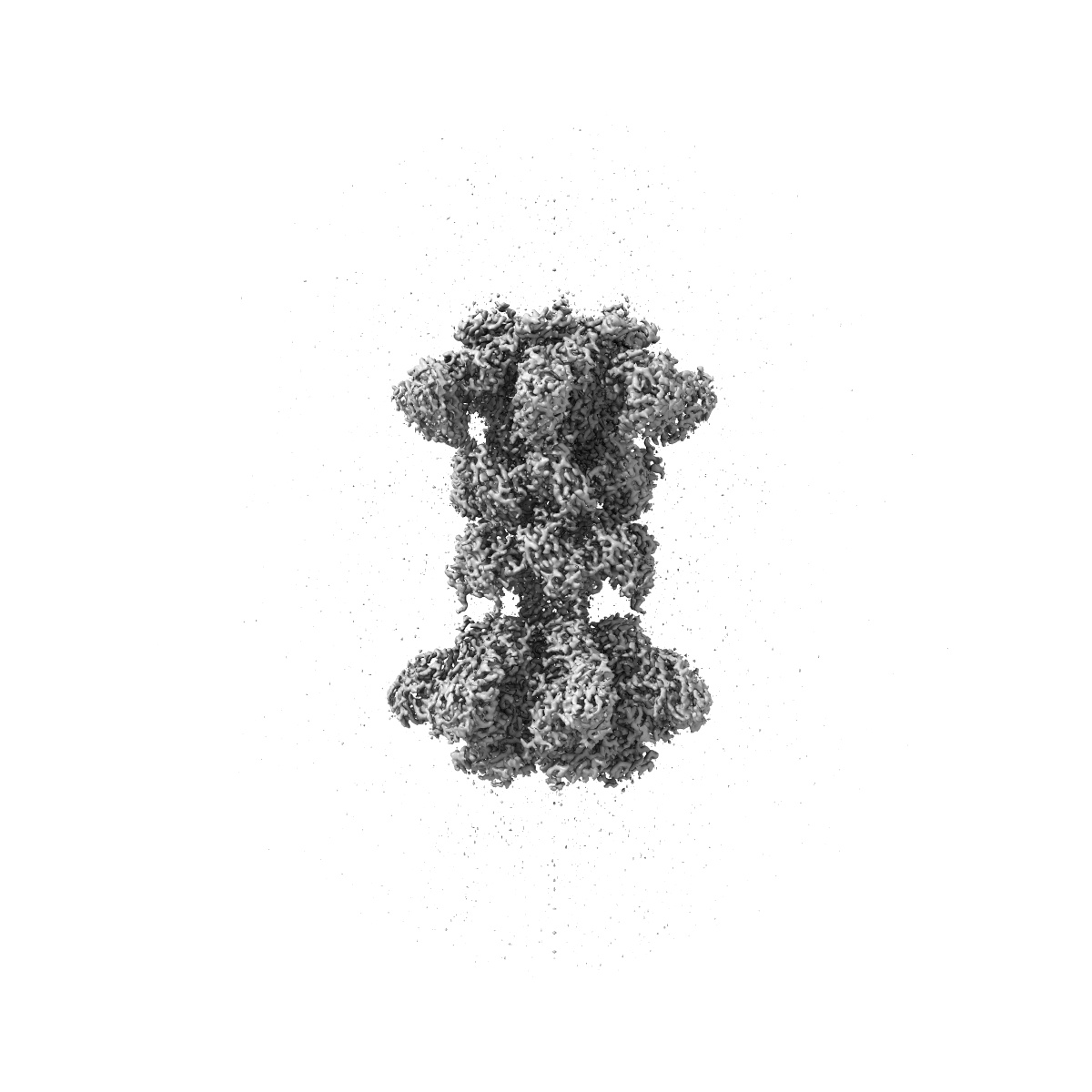
Clostridium difficile binary toxin translocase CDTb in asymmetric tetradecamer conformation
EMD-20926
Single-particle2.8 Å
 Deposition: 05/11/2019
Deposition: 05/11/2019Map released: 22/01/2020
Last modified: 06/03/2024
Sample Organism:
Clostridioides difficile
Sample: Tetradecamer of CDTb
Fitted models: 6uwr (Avg. Q-score: 0.594)
Raw data: EMPIAR-11703
Deposition Authors: Xu X ,
Pozharski E
,
Pozharski E
Sample: Tetradecamer of CDTb
Fitted models: 6uwr (Avg. Q-score: 0.594)
Raw data: EMPIAR-11703
Deposition Authors: Xu X
 ,
Pozharski E
,
Pozharski E
Structure of the cell-binding component of theClostridium difficilebinary toxin reveals a di-heptamer macromolecular assembly.
Xu X ,
Godoy-Ruiz R ,
Adipietro KA,
Peralta C ,
Ben-Hail D ,
Varney KM,
Cook ME ,
Roth BM,
Wilder PT,
Cleveland T ,
Grishaev A,
Neu HM,
Michel SLJ ,
Yu W ,
Beckett D,
Rustandi RR,
Lancaster C,
Loughney JW,
Kristopeit A,
Christanti S,
Olson JW,
MacKerell AD,
Georges AD,
Pozharski E,
Weber DJ
(2020) PNAS , 117 , 1049 - 1058
,
Godoy-Ruiz R ,
Adipietro KA,
Peralta C ,
Ben-Hail D ,
Varney KM,
Cook ME ,
Roth BM,
Wilder PT,
Cleveland T ,
Grishaev A,
Neu HM,
Michel SLJ ,
Yu W ,
Beckett D,
Rustandi RR,
Lancaster C,
Loughney JW,
Kristopeit A,
Christanti S,
Olson JW,
MacKerell AD,
Georges AD,
Pozharski E,
Weber DJ
(2020) PNAS , 117 , 1049 - 1058
Abstract:
Targeting Clostridium difficile infection is challenging because treatment options are limited, and high recurrence rates are common. One reason for this is that hypervirulent C. difficile strains often have a binary toxin termed the C. difficile toxin, in addition to the enterotoxins TsdA and TsdB. The C. difficile toxin has an enzymatic component, termed CDTa, and a pore-forming or delivery subunit termed CDTb. CDTb was characterized here using a combination of single-particle cryoelectron microscopy, X-ray crystallography, NMR, and other biophysical methods. In the absence of CDTa, 2 di-heptamer structures for activated CDTb (1.0 MDa) were solved at atomic resolution, including a symmetric (SymCDTb; 3.14 Å) and an asymmetric form (AsymCDTb; 2.84 Å). Roles played by 2 receptor-binding domains of activated CDTb were of particular interest since the receptor-binding domain 1 lacks sequence homology to any other known toxin, and the receptor-binding domain 2 is completely absent in other well-studied heptameric toxins (i.e., anthrax). For AsymCDTb, a Ca2+ binding site was discovered in the first receptor-binding domain that is important for its stability, and the second receptor-binding domain was found to be critical for host cell toxicity and the di-heptamer fold for both forms of activated CDTb. Together, these studies represent a starting point for developing structure-based drug-design strategies to target the most severe strains of C. difficile.
Targeting Clostridium difficile infection is challenging because treatment options are limited, and high recurrence rates are common. One reason for this is that hypervirulent C. difficile strains often have a binary toxin termed the C. difficile toxin, in addition to the enterotoxins TsdA and TsdB. The C. difficile toxin has an enzymatic component, termed CDTa, and a pore-forming or delivery subunit termed CDTb. CDTb was characterized here using a combination of single-particle cryoelectron microscopy, X-ray crystallography, NMR, and other biophysical methods. In the absence of CDTa, 2 di-heptamer structures for activated CDTb (1.0 MDa) were solved at atomic resolution, including a symmetric (SymCDTb; 3.14 Å) and an asymmetric form (AsymCDTb; 2.84 Å). Roles played by 2 receptor-binding domains of activated CDTb were of particular interest since the receptor-binding domain 1 lacks sequence homology to any other known toxin, and the receptor-binding domain 2 is completely absent in other well-studied heptameric toxins (i.e., anthrax). For AsymCDTb, a Ca2+ binding site was discovered in the first receptor-binding domain that is important for its stability, and the second receptor-binding domain was found to be critical for host cell toxicity and the di-heptamer fold for both forms of activated CDTb. Together, these studies represent a starting point for developing structure-based drug-design strategies to target the most severe strains of C. difficile.