{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
EMD-26607
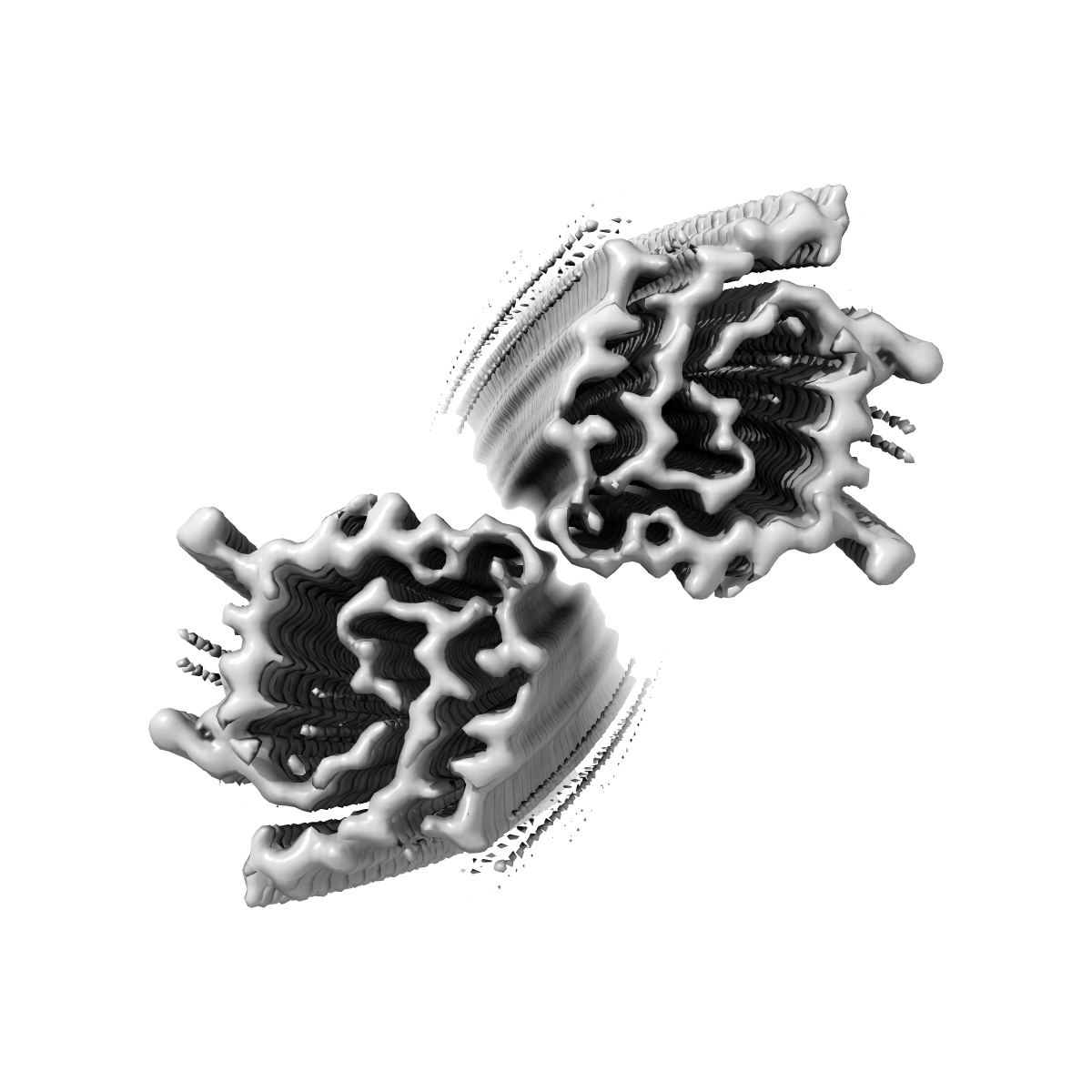
Structure of Type I Prion filaments from Gerstmann-Straussler-Scheinker disease
EMD-26607
Helical reconstruction3.29 Å
 Deposition: 07/04/2022
Deposition: 07/04/2022Map released: 27/07/2022
Last modified: 27/03/2024
Sample Organism:
Homo sapiens
Sample: Type I Prion protein filament from Gerstmann-Straussler-Scheinker disease
Fitted models: 7umq (Avg. Q-score: 0.481)
Deposition Authors: Ozcan KO, Hoq MR, Bharath SR, Jiang W
Sample: Type I Prion protein filament from Gerstmann-Straussler-Scheinker disease
Fitted models: 7umq (Avg. Q-score: 0.481)
Deposition Authors: Ozcan KO, Hoq MR, Bharath SR, Jiang W
Cryo-EM structures of prion protein filaments from Gerstmann-Straussler-Scheinker disease.
Hallinan GI,
Ozcan KA  ,
Hoq MR,
Cracco L,
Vago FS,
Bharath SR,
Li D,
Jacobsen M,
Doud EH,
Mosley AL,
Fernandez A ,
Garringer HJ ,
Jiang W,
Ghetti B,
Vidal R
,
Hoq MR,
Cracco L,
Vago FS,
Bharath SR,
Li D,
Jacobsen M,
Doud EH,
Mosley AL,
Fernandez A ,
Garringer HJ ,
Jiang W,
Ghetti B,
Vidal R
(2022) Acta Neuropathol , 144 , 509 - 520
 ,
Hoq MR,
Cracco L,
Vago FS,
Bharath SR,
Li D,
Jacobsen M,
Doud EH,
Mosley AL,
Fernandez A ,
Garringer HJ ,
Jiang W,
Ghetti B,
Vidal R
,
Hoq MR,
Cracco L,
Vago FS,
Bharath SR,
Li D,
Jacobsen M,
Doud EH,
Mosley AL,
Fernandez A ,
Garringer HJ ,
Jiang W,
Ghetti B,
Vidal R
(2022) Acta Neuropathol , 144 , 509 - 520
Abstract:
Prion protein (PrP) aggregation and formation of PrP amyloid (APrP) are central events in the pathogenesis of prion diseases. In the dominantly inherited prion protein amyloidosis known as Gerstmann-Sträussler-Scheinker (GSS) disease, plaques made of PrP amyloid are present throughout the brain. The c.593t > c mutation in the prion protein gene (PRNP) results in a phenylalanine to serine amino acid substitution at PrP residue 198 (F198S) and causes the most severe amyloidosis among GSS variants. It has been shown that neurodegeneration in this disease is associated with the presence of extracellular APrP plaques and neuronal intracytoplasmic Tau inclusions, that have been shown to contain paired helical filaments identical to those found in Alzheimer disease. Using cryogenic electron microscopy (cryo-EM), we determined for the first time the structures of filaments of human APrP, isolated post-mortem from the brain of two symptomatic PRNP F198S mutation carriers. We report that in GSS (F198S) APrP filaments are composed of dimeric, trimeric and tetrameric left-handed protofilaments with their protomers sharing a common protein fold. The protomers in the cross-β spines consist of 62 amino acids and span from glycine 80 to phenylalanine 141, adopting a previously unseen spiral fold with a thicker outer layer and a thinner inner layer. Each protomer comprises nine short β-strands, with the β1 and β8 strands, as well as the β4 and β9 strands, forming a steric zipper. The data obtained by cryo-EM provide insights into the structural complexity of the PrP filament in a dominantly inherited human PrP amyloidosis. The novel findings highlight the urgency of extending our knowledge of the filaments' structures that may underlie distinct clinical and pathologic phenotypes of human neurodegenerative diseases.
Prion protein (PrP) aggregation and formation of PrP amyloid (APrP) are central events in the pathogenesis of prion diseases. In the dominantly inherited prion protein amyloidosis known as Gerstmann-Sträussler-Scheinker (GSS) disease, plaques made of PrP amyloid are present throughout the brain. The c.593t > c mutation in the prion protein gene (PRNP) results in a phenylalanine to serine amino acid substitution at PrP residue 198 (F198S) and causes the most severe amyloidosis among GSS variants. It has been shown that neurodegeneration in this disease is associated with the presence of extracellular APrP plaques and neuronal intracytoplasmic Tau inclusions, that have been shown to contain paired helical filaments identical to those found in Alzheimer disease. Using cryogenic electron microscopy (cryo-EM), we determined for the first time the structures of filaments of human APrP, isolated post-mortem from the brain of two symptomatic PRNP F198S mutation carriers. We report that in GSS (F198S) APrP filaments are composed of dimeric, trimeric and tetrameric left-handed protofilaments with their protomers sharing a common protein fold. The protomers in the cross-β spines consist of 62 amino acids and span from glycine 80 to phenylalanine 141, adopting a previously unseen spiral fold with a thicker outer layer and a thinner inner layer. Each protomer comprises nine short β-strands, with the β1 and β8 strands, as well as the β4 and β9 strands, forming a steric zipper. The data obtained by cryo-EM provide insights into the structural complexity of the PrP filament in a dominantly inherited human PrP amyloidosis. The novel findings highlight the urgency of extending our knowledge of the filaments' structures that may underlie distinct clinical and pathologic phenotypes of human neurodegenerative diseases.