{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
EMD-4591
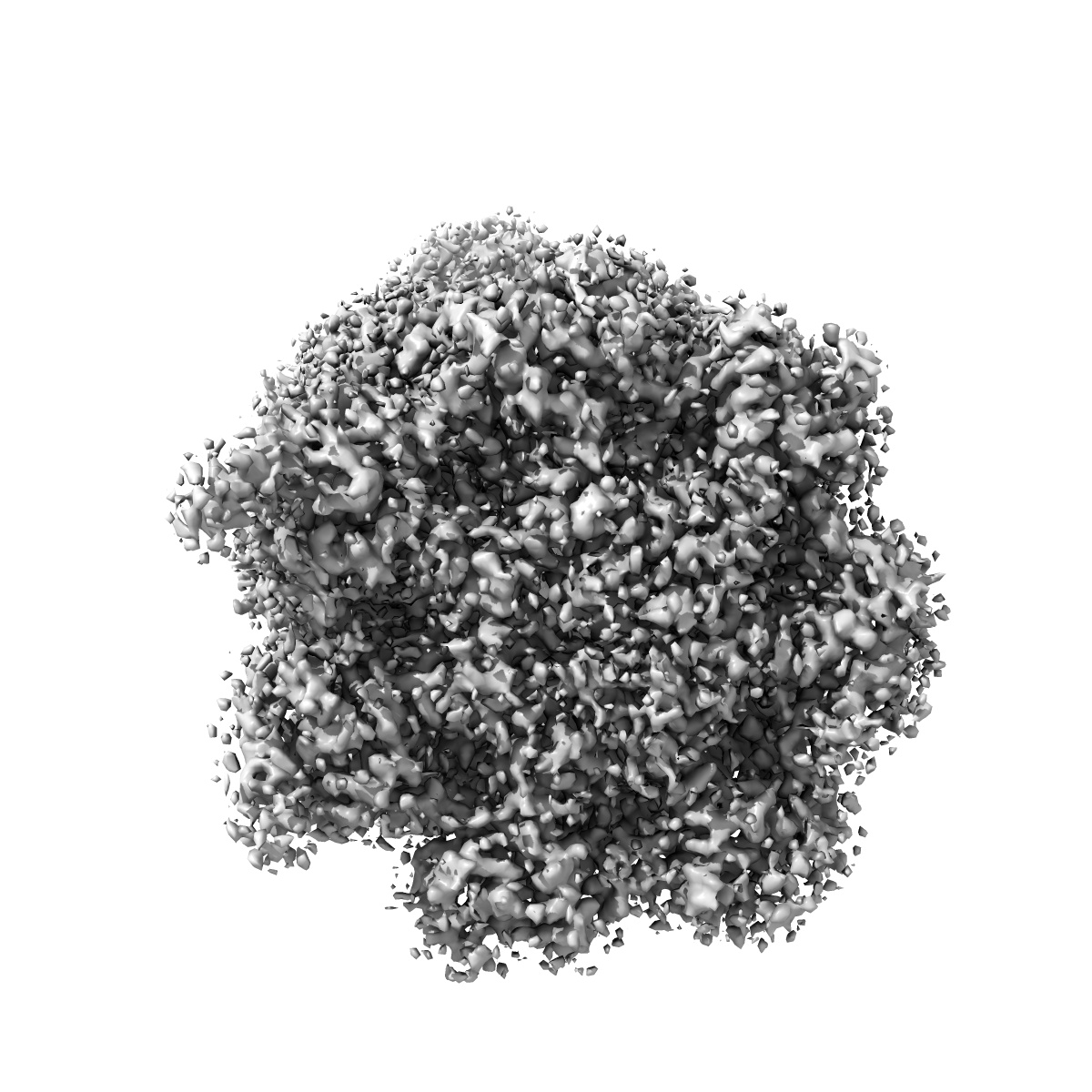
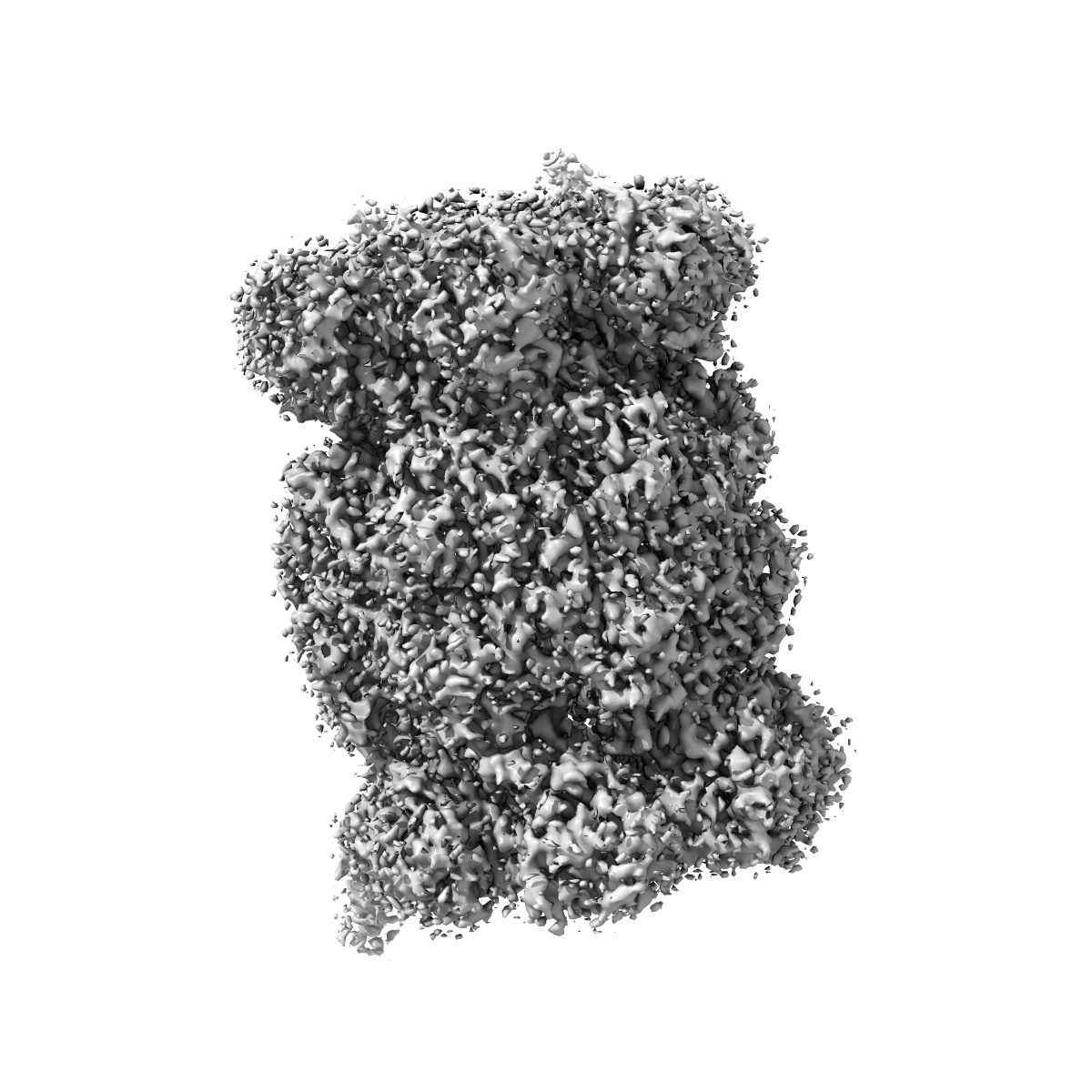
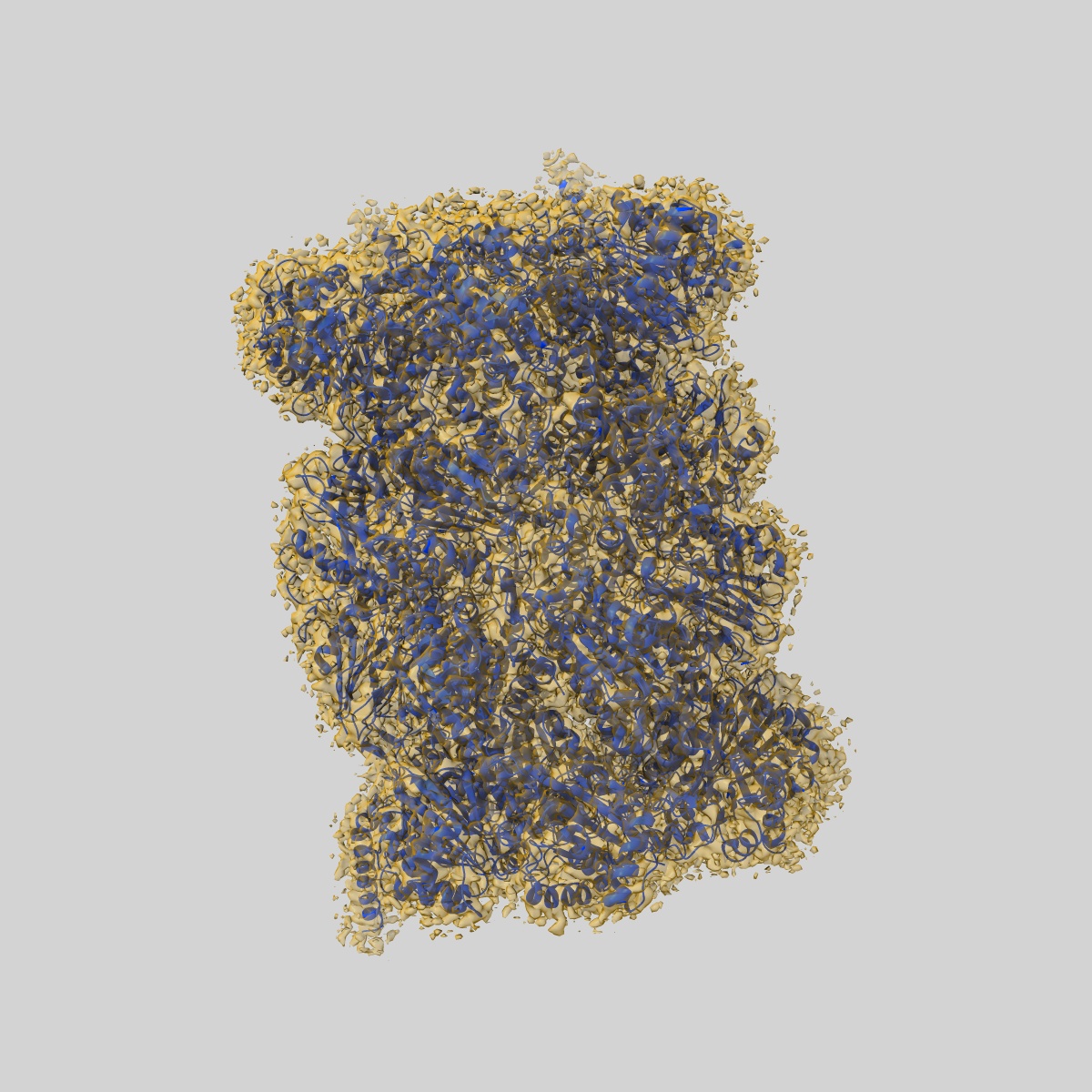
Leishmania tarentolae proteasome 20S subunit apo structure
EMD-4591
Single-particle3.3 Å
 Deposition: 01/02/2019
Deposition: 01/02/2019Map released: 17/04/2019
Last modified: 06/11/2024
Sample Organism:
Leishmania tarentolae
Sample: Proteasome 20S subunit
Fitted models: 6qm8 (Avg. Q-score: 0.516)
Deposition Authors: Goswami P, Rowland P
Sample: Proteasome 20S subunit
Fitted models: 6qm8 (Avg. Q-score: 0.516)
Deposition Authors: Goswami P, Rowland P
Preclinical candidate for the treatment of visceral leishmaniasis that acts through proteasome inhibition.
Wyllie S  ,
Brand S ,
Thomas M,
De Rycker M ,
Chung CW,
Pena I,
Bingham RP,
Bueren-Calabuig JA,
Cantizani J,
Cebrian D,
Craggs PD ,
Ferguson L,
Goswami P,
Hobrath J,
Howe J,
Jeacock L,
Ko EJ ,
Korczynska J,
MacLean L,
Manthri S,
Martinez MS,
Mata-Cantero L,
Moniz S,
Nuhs A,
Osuna-Cabello M,
Pinto E,
Riley J,
Robinson S,
Rowland P,
Simeons FRC,
Shishikura Y,
Spinks D,
Stojanovski L,
Thomas J,
Thompson S,
Viayna Gaza E,
Wall RJ ,
Zuccotto F ,
Horn D ,
Ferguson MAJ,
Fairlamb AH ,
Fiandor JM,
Martin J,
Gray DW ,
Miles TJ,
Gilbert IH ,
Read KD,
Marco M ,
Wyatt PG
,
Brand S ,
Thomas M,
De Rycker M ,
Chung CW,
Pena I,
Bingham RP,
Bueren-Calabuig JA,
Cantizani J,
Cebrian D,
Craggs PD ,
Ferguson L,
Goswami P,
Hobrath J,
Howe J,
Jeacock L,
Ko EJ ,
Korczynska J,
MacLean L,
Manthri S,
Martinez MS,
Mata-Cantero L,
Moniz S,
Nuhs A,
Osuna-Cabello M,
Pinto E,
Riley J,
Robinson S,
Rowland P,
Simeons FRC,
Shishikura Y,
Spinks D,
Stojanovski L,
Thomas J,
Thompson S,
Viayna Gaza E,
Wall RJ ,
Zuccotto F ,
Horn D ,
Ferguson MAJ,
Fairlamb AH ,
Fiandor JM,
Martin J,
Gray DW ,
Miles TJ,
Gilbert IH ,
Read KD,
Marco M ,
Wyatt PG
(2019) PNAS , 116 , 9318 - 9323
 ,
Brand S ,
Thomas M,
De Rycker M ,
Chung CW,
Pena I,
Bingham RP,
Bueren-Calabuig JA,
Cantizani J,
Cebrian D,
Craggs PD ,
Ferguson L,
Goswami P,
Hobrath J,
Howe J,
Jeacock L,
Ko EJ ,
Korczynska J,
MacLean L,
Manthri S,
Martinez MS,
Mata-Cantero L,
Moniz S,
Nuhs A,
Osuna-Cabello M,
Pinto E,
Riley J,
Robinson S,
Rowland P,
Simeons FRC,
Shishikura Y,
Spinks D,
Stojanovski L,
Thomas J,
Thompson S,
Viayna Gaza E,
Wall RJ ,
Zuccotto F ,
Horn D ,
Ferguson MAJ,
Fairlamb AH ,
Fiandor JM,
Martin J,
Gray DW ,
Miles TJ,
Gilbert IH ,
Read KD,
Marco M ,
Wyatt PG
,
Brand S ,
Thomas M,
De Rycker M ,
Chung CW,
Pena I,
Bingham RP,
Bueren-Calabuig JA,
Cantizani J,
Cebrian D,
Craggs PD ,
Ferguson L,
Goswami P,
Hobrath J,
Howe J,
Jeacock L,
Ko EJ ,
Korczynska J,
MacLean L,
Manthri S,
Martinez MS,
Mata-Cantero L,
Moniz S,
Nuhs A,
Osuna-Cabello M,
Pinto E,
Riley J,
Robinson S,
Rowland P,
Simeons FRC,
Shishikura Y,
Spinks D,
Stojanovski L,
Thomas J,
Thompson S,
Viayna Gaza E,
Wall RJ ,
Zuccotto F ,
Horn D ,
Ferguson MAJ,
Fairlamb AH ,
Fiandor JM,
Martin J,
Gray DW ,
Miles TJ,
Gilbert IH ,
Read KD,
Marco M ,
Wyatt PG
(2019) PNAS , 116 , 9318 - 9323
Abstract:
Visceral leishmaniasis (VL), caused by the protozoan parasites Leishmania donovani and Leishmania infantum, is one of the major parasitic diseases worldwide. There is an urgent need for new drugs to treat VL, because current therapies are unfit for purpose in a resource-poor setting. Here, we describe the development of a preclinical drug candidate, GSK3494245/DDD01305143/compound 8, with potential to treat this neglected tropical disease. The compound series was discovered by repurposing hits from a screen against the related parasite Trypanosoma cruzi Subsequent optimization of the chemical series resulted in the development of a potent cidal compound with activity against a range of clinically relevant L. donovani and L. infantum isolates. Compound 8 demonstrates promising pharmacokinetic properties and impressive in vivo efficacy in our mouse model of infection comparable with those of the current oral antileishmanial miltefosine. Detailed mode of action studies confirm that this compound acts principally by inhibition of the chymotrypsin-like activity catalyzed by the β5 subunit of the L. donovani proteasome. High-resolution cryo-EM structures of apo and compound 8-bound Leishmania tarentolae 20S proteasome reveal a previously undiscovered inhibitor site that lies between the β4 and β5 proteasome subunits. This induced pocket exploits β4 residues that are divergent between humans and kinetoplastid parasites and is consistent with all of our experimental and mutagenesis data. As a result of these comprehensive studies and due to a favorable developability and safety profile, compound 8 is being advanced toward human clinical trials.
Visceral leishmaniasis (VL), caused by the protozoan parasites Leishmania donovani and Leishmania infantum, is one of the major parasitic diseases worldwide. There is an urgent need for new drugs to treat VL, because current therapies are unfit for purpose in a resource-poor setting. Here, we describe the development of a preclinical drug candidate, GSK3494245/DDD01305143/compound 8, with potential to treat this neglected tropical disease. The compound series was discovered by repurposing hits from a screen against the related parasite Trypanosoma cruzi Subsequent optimization of the chemical series resulted in the development of a potent cidal compound with activity against a range of clinically relevant L. donovani and L. infantum isolates. Compound 8 demonstrates promising pharmacokinetic properties and impressive in vivo efficacy in our mouse model of infection comparable with those of the current oral antileishmanial miltefosine. Detailed mode of action studies confirm that this compound acts principally by inhibition of the chymotrypsin-like activity catalyzed by the β5 subunit of the L. donovani proteasome. High-resolution cryo-EM structures of apo and compound 8-bound Leishmania tarentolae 20S proteasome reveal a previously undiscovered inhibitor site that lies between the β4 and β5 proteasome subunits. This induced pocket exploits β4 residues that are divergent between humans and kinetoplastid parasites and is consistent with all of our experimental and mutagenesis data. As a result of these comprehensive studies and due to a favorable developability and safety profile, compound 8 is being advanced toward human clinical trials.