- Course overview
- Search within this course
- Pfam entry types
- What are profile hidden Markov models?
- Modelling in Pfam
- Grouping Pfam entries into Clans
- Summary
- Get help and support on Pfam
- Your feedback
- References
![]()
All materials are free cultural works licensed under a Creative Commons Attribution 4.0 International (CC BY 4.0) license, except where further licensing details are provided.
What are profile hidden Markov models?
They are one of the computational algorithms used for predicting protein structure and function, identifies significant protein sequence similarities allowing the detection of homologs and consequently the transfer of information, i.e. sequence homology-based inference of knowledge. In this section we will describe the algorithm used to create Pfam entries: profile hidden Markov models (HMMs).
Profile HMMs are probabilistic models that encapsulate the evolutionary changes that have occurred in a set of related sequences (i.e. a multiple sequence alignment). To do so, they capture position-specific information about how conserved each amino acid is in each column of the alignment, see Figure 2.
The model also captures important information such as the varying degree to which gaps and insertions have occurred. Unlike other sequence homology detection algorithms, profile HMMs use position dependent gap penalties and substitution probabilities which better reflect biological reality [1].
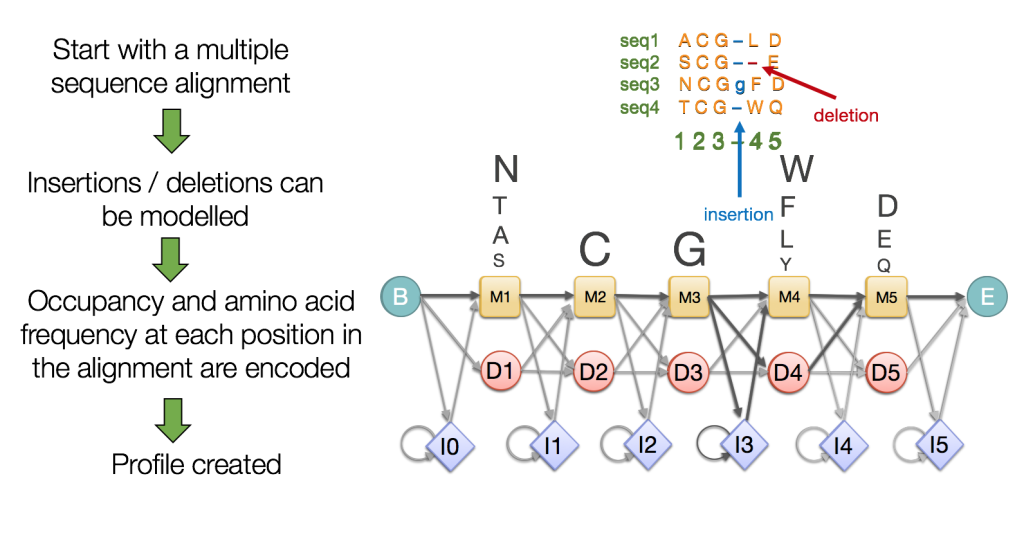
The boxes in yellow are the match states (M). In the M state the probability distribution is the frequency of the amino acids in that position. The row of diamond shaped states are insert states (I) which are used to model highly variable regions in the alignment. The circular states are delete states (D). These are called silent states since they do not match any residues, and they are there merely to make it possible to jump over one or more columns in the alignment.The final probabilistic model conveys the estimation of the observed frequencies of the amino acids in each position, as well as the transitions between the amino acids derived from the observed occupancy of each position in a multiple sequence alignment.